In solid-state physics, the tight binding model (or TB model) is an approach to the calculation of electronic band structure using an approximate set of wave functions based upon superposition of wave functions for isolated atoms located at each atomic site. The method is closely related to the linear combination of atomic orbitals molecular orbital method used for molecules. Tight binding calculates the ground state electronic energy and position of bandgaps for a molecule.
This approximation could be considered the analog of the LCAO-MO (Linear Combination of Atomic Orbital- Molecular Orbital) approach. The idea of a molecular orbital was advanced by Friedrich Hund and Robert Mulliken from 1926 through 1928. The LCAO method for approximating molecular orbitals was introduced in 1928 by B. N. Finklestein and G. E. Horowitz, while the LCAO for solids was developed by Felix Bloch, as part of his doctoral dissertation in 1928, concurrently with and independent of the LCAO-MO approach. A much simpler interpolation scheme for approximating the electronic band structure, the tight-binding method was originally conceived in 1954 by John Clarke Slater and George Fred Koster. Hence, it is sometimes referred to as the SK method. As in the LCAO-MO approach, the atomic locations can be specified arbitrarily (or additional calculations can be done to find the atomic positions), so the method can be applied to non-crystalline materials. However, the most common applications are to crystalline materials where the atomic positions are located on a periodic space lattice of sites. With the tight-binding method, the electronic band structure of a solid is interpolated over the entire Brillouin zone by fitting to first-principles calculations carried out at high-symmetry points.
In this approach, interactions between different atomic sites are considered as perturbations. There exists several kinds of interactions we must consider. The crystal Hamiltonian is only approximately a sum of atomic Hamiltonians located at different sites and atomic wave functions overlap adjacent atomic sites in the crystal, and so are not accurate representations of the exact wave function. There are further explanations in the next section with some mathematical expressions.
Recently, in the research about strongly correlated material, the tight binding approach is basic approximation because highly localized electrons like 3-d transition metal electrons sometimes indicate strongly correlated behaviors. In this case, the role of electron-electron interaction must be considered using the many-body physics description.
The tight-binding model is typically used for calculations of electronic band structure and band gaps in the static regime. However, in combination with other methods such as the random phase approximation (RPA) model, the dynamic response of systems may also be studied.
Formulation
We introduce the atomic orbitals φm( r ), which are eigenfunctions of the Hamiltonian Hat of a single isolated atom. When the atom is placed in a crystal, this atomic wave function overlaps adjacent atomic sites, and so are not true eigenfunctions of the crystal Hamiltonian. The overlap is less when electrons are tightly bound, which is the source of the descriptor "tight-binding". Any corrections to the atomic potential ΔU required to obtain the true Hamiltonian H of the system, are assumed small:
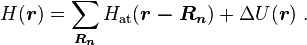
A solution ψ(r) to the time-independent single electron Schrödinger equation is then approximated as a linear combination of atomic orbitals φm( r − Rn ):
 ,
,
where m refers to the m-th atomic energy level and Rn locates an atomic site in the crystal lattice.
The translational symmetry of the crystal implies the wave function under translation can change only by a phase factor:
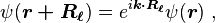
where k is the wave vector of the wave function. Consequently, the coefficients satisfy
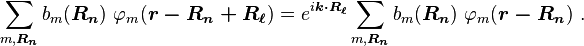
By substituting Rp = Rn − Rℓ, we find

or

Normalizing the wave function to unity:
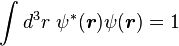
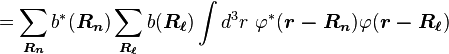

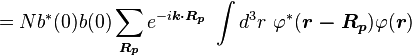
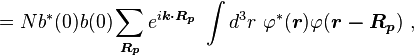
so the normalization sets b(0) as
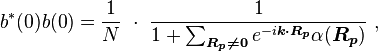
where α (Rp ) are the atomic overlap integrals, which frequently are neglected resulting in[1]
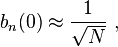
and
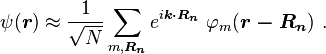
Using the tight binding form for the wave function, and assuming only the m-th atomic energy level is important for the m-th energy band, the Bloch energies 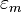 are of the form
are of the form
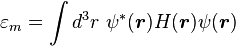
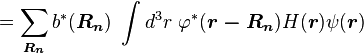
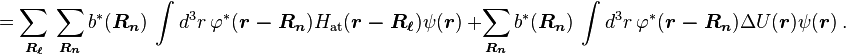
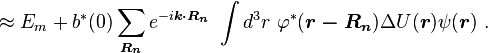
Here terms involving the atomic Hamiltonian at sites other than where it is centered are neglected. The energy then becomes

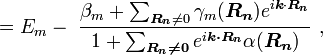
where Em is the energy of the m-th atomic level,
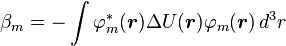 ,
,

and
 ,
,
are the overlap integrals.
One-dimensional example
Here the tight binding model is illustrated for a string of atoms in a straight line with spacing a between atomic sites. Denote the translation operator τ, which satisfies the property:

Here, the state ket  represents a particular choice of atomic orbital (for example, an s- or p- orbital from some shell of orbitals) located at the site Rn = n a in the lattice with lattice constant a. Because the Hamiltonian H is invariant under the operation τ(a), we have commutation relation
represents a particular choice of atomic orbital (for example, an s- or p- orbital from some shell of orbitals) located at the site Rn = n a in the lattice with lattice constant a. Because the Hamiltonian H is invariant under the operation τ(a), we have commutation relation
![[H,\ \tau{(a)}]=0](http://upload.wikimedia.org/math/2/e/1/2e14f00b2029d9ea47c4d79f02380be7.png) .
.
This commutation relation implies the Hamiltonian operator H and translation operator τ(a) can be simultaneously diagonalized.
To find approximate eigenstates of the Hamiltonian, we can use a linear combination of the atomic orbitals
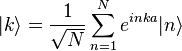
where N = total number of sites and k is a real parameter with  . (This wave function is normalized to unity by the leading factor 1/√N provided overlap of atomic wave functions is ignored.) If we apply the lattice translation operator τ(a) to this state
. (This wave function is normalized to unity by the leading factor 1/√N provided overlap of atomic wave functions is ignored.) If we apply the lattice translation operator τ(a) to this state 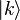 , this state is found to be an eigenstate of this operator:
, this state is found to be an eigenstate of this operator:

Assuming only nearest neighbor overlap (that is, tight binding), the only non-zero matrix elements of the Hamiltonian can be expressed as


The energy E0 is approximately the atomic energy level corresponding to the chosen atomic orbital if H at site Rn = n a is approximately Hat at that site. We can derive the energy of the state using the above equation:
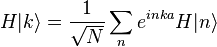
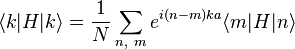
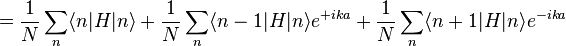

where, for example,
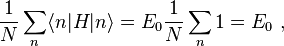
and
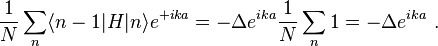
Thus the energy of this state can be represented in the familiar form of the energy dispersion:
 .
.
This example is readily extended to three dimensions, for example, to a body-centered cubic or face-centered cubic lattice by introducing the nearest neighbor vector locations in place of simply n a.Likewise, the method can be extended to multiple bands using multiple different atomic orbitals at each site. The general formulation above shows how these extensions can be accomplished.
Connection to Wannier functions
Bloch wave functions describe the electronic states in a periodic crystal lattice. Bloch functions can be represented as a Fourier series
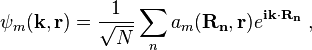
where Rn denotes an atomic site in a periodic crystal lattice, k is the wave vector of the Bloch wave, r is the electron position, m is the band index, and the sum is over all N atomic sites. The Bloch wave is an exact eigensolution for the wave function of an electron in a periodic crystal potential corresponding to an energy Em (k), and is spread over the entire crystal volume.
Using the Fourier transform analysis, a spatially localized wave function for the m-th energy band can be derived from this Bloch wave:

These real space wave functions  are called Wannier functions, and are fairly closely localized to the atomic site Rn. Of course, if we have exact Wannier functions, the exact Bloch functions can be derived using the inverse Fourier transform.
are called Wannier functions, and are fairly closely localized to the atomic site Rn. Of course, if we have exact Wannier functions, the exact Bloch functions can be derived using the inverse Fourier transform.
However it is not easy to calculate directly either Bloch functions or Wannier functions. An approximate approach is necessary in the calculation of electronic structures of solids. If we consider the extreme case of isolated atoms, the Wannier function would become an isolated atomic orbital. That limit suggests the choice of an atomic wave function as an approximate form for the Wannier function, the so-called tight binding approximation.
Second quantization
Modern explanations of electronic structure like t-J model and Hubbard model are based on tight binding model. If we introduce second quantization formalism, it is clear to understand the concept of tight binding model.
Using the atomic orbital as a basis state, we can establish the second quantization Hamiltonian operator in tight binding model.
 ,
,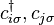 - creation and annihilation operators
- creation and annihilation operators
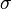 - spin polarization
- spin polarization
 - hopping integral
- hopping integral
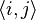 -nearest neighbor index
-nearest neighbor index
Here, hopping integral corresponds to the transfer integral 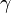 in tight binding model. Considering extreme cases of
in tight binding model. Considering extreme cases of 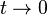 , it is impossible for electron to hop into neighboring sites. This case is the isolated atomic system. If the hopping term is turned on (
, it is impossible for electron to hop into neighboring sites. This case is the isolated atomic system. If the hopping term is turned on (0" src="http://upload.wikimedia.org/math/c/4/2/c42a280003a64774acf91ad359d199bc.png">) electrons can stay in both sites lowering their kinetic energy.
In the strongly correlated electron system, it is necessary to consider the electron-electron interaction. This term can be written in
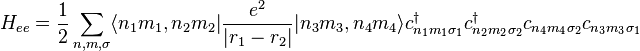
This interaction Hamiltonian includes direct Coulomb interaction energy and exchange interaction energy between electrons. There are several novel physics induced from this electron-electron interaction energy, such as metal-insulator transitions (MIT), high-temperature superconductivity, and several quantum phase transitions.
No hay comentarios:
Publicar un comentario